Sequencing Solutions
Genome-wide detection and phasing of genetic and epigenetic variants from a single library prep- Two Features at One Go
Detects distinct regional epigenetic patterns
Accesses methylation in the full genome
Identifies allele-specific methylation

Whole Genome Sequencing
Gain an unbiased, base-by-base view of the entire genome. WGS detects single nucleotide variants, insertions /deletions, copy number variations (CNVs) and structural variants. It’s widely used in population genetics, rare disease research, evolutionary studies, and complex trait mapping.
Whole Exome Sequencing (WES)
Focus on the protein-coding regions that account for ~1–2% of the genome but harbor ~85% of disease-causing mutations. WES is a cost-effective strategy for identifying pathogenic variants, especially in clinical diagnostics and hereditary disease research.
Metagenomics
The study of genetic material recovered directly from environmental samples, such as soil, water, or the human microbiome, without the need for culturing individual organisms. Unlike traditional genomics, which focuses on a single organism’s DNA, metagenomics provides a comprehensive view of entire microbial communities
Sanger Sequencing
Sanger Sequencing is a classical method of DNA sequencing developed by Frederick Sanger in 1977. It is also known as the chain-termination method and is widely used for accurate, small-scale DNA sequencing (e.g., verifying cloned DNA, detecting mutations, sequencing plasmids or PCR products).
Single-cell Sequencing
Dissect cellular heterogeneity by profiling gene expression in thousands of individual cells. Ideal for understanding tissue composition, immune landscapes, stem cell differentiation, or tumor microenvironments. Supports droplet-based and plate-based protocols.
Epigenomics
Epigenomics is the study of the chemical changes to DNA and histones that regulate gene activity without altering the underlying genetic sequence. These changes, such as DNA methylation and histone modification, can influence gene expression, impacting cellular processes like development, aging, and disease. Epigenetic marks can be passed down to offspring, affecting inheritance and evolution, and are influenced by environmental factors, lifestyle, and external stresses.
Spatial Sequencing
Spatial sequencing bridges the gap between single-cell genomics and tissue context by preserving the physical location of cells while analyzing their molecular profiles. This cutting-edge technique combines high-resolution imaging with NGS or in situ sequencing to reveal how gene expression varies across tissue microenvironments—critical for understanding tumor heterogeneity, immune cell interactions, and developmental biology.
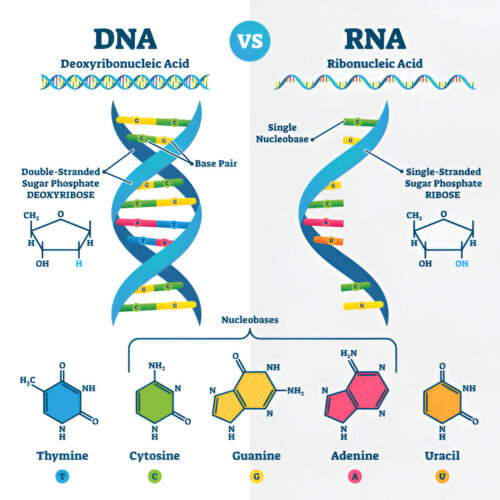
RNA Sequencing
Unlock transcriptomic data to study gene expression, differential expression, and alternative splicing. RNA-Seq enables comprehensive insights into coding and non-coding RNAs, revealing functional elements of the genome and offering a dynamic view of cellular states across conditions or tissues.
Pre-Med Library Sequencing
Pre-Med Library Sequencing refers to targeted sequencing of a curated panel of genes known to be associated with a broad range of inherited conditions and actionable clinical outcomes. Designed for preventive healthcare, this approach enables early detection of genetic variants linked to conditions such as cancer, cardiovascular disorders, neurological diseases, and pharmacogenomic responses.
Isolation of DNA/RNA
Isolation of DNA/RNA is a fundamental step in molecular biology, enabling the extraction of high-quality genetic material from biological samples such as blood, tissues, or cells. Accurate and efficient isolation is critical for downstream applications including sequencing, PCR, gene expression analysis, and diagnostic testing.
Whole Genome sequencing
Whole-genome sequencing (WGS) is a powerful technique that decodes the complete DNA of an organism. It plays a key role in identifying genetic disorders, cancer mutations, and tracking disease outbreaks. Thanks to advances in next-generation sequencing (NGS), WGS is now faster and more affordable, making it useful not only for human genomes but also for studying plants, animals, and microbes in agriculture and research.
Long-read sequencing (e.g., PacBio HiFi) generates ultra-long, accurate reads that resolve complex genomic regions, full-length transcripts, and structural variations. It enables:
- Telomere-to-telomere genome assemblies
- Accurate isoform detection (Iso-Seq)
- Direct epigenetic detection
- Strain-level microbiome analysis
LRS is transforming rare disease diagnosis, cancer genomics, and functional annotation by providing a more complete view of the genome and transcriptome.
Whole Exome Sequencing
Whole exome sequencing (WES) focuses on the protein-coding regions of the genome, where most disease-related mutations occur. It’s a cost-effective method for identifying potential genetic causes of disease. In addition, techniques like copy number variation (CNV) and structural variant (SV) analysis help detect genomic alterations that affect gene expression, offering deeper insights into the genetic basis of various disorders.

Metagenomics
The study of genetic material recovered directly from environmental samples, such as soil, water, or the human microbiome, without the need for culturing individual organisms. Unlike traditional genomics, which focuses on a single organism’s DNA, metagenomics provides a comprehensive view of entire microbial communities, revealing their diversity, functions, and interactions.
ITS sequencing targets variable genomic regions to provide precise fungal and eukaryotic identification. It is widely used to study diversity in environmental, clinical, and industrial samples—offering species-level resolution where 16S falls short. A gold standard for detecting fungi, molds, and protists in microbiome research
Single Cell Sequencing
Single-cell sequencing analyzes the genetic material of individual cells, uncovering hidden diversity missed by bulk methods. It helps identify rare cell types, trace development, and reveal disease mechanisms, advancing fields like cancer research, neurobiology, and immunotherapy.

Epigenomics
Epigenomics is the study of the chemical changes to DNA and histones that regulate gene activity without altering the underlying genetic sequence. These changes, such as DNA methylation and histone modification, can influence gene expression, impacting cellular processes like development, aging, and disease. Epigenetic marks can be passed down to offspring, affecting inheritance and evolution, and are influenced by environmental factors, lifestyle, and external stresses.
ChIP-sequencing (Chromatin Immunoprecipitation Sequencing) is a powerful technique used to analyze protein-DNA interactions and study the epigenome. It combines chromatin immunoprecipitation (ChIP) with next-generation sequencing (NGS) to identify binding sites of DNA-associated proteins (like transcription factors, histones, or other chromatin modifiers) across the entire genome.

Spatial Sequencing
Spatial sequencing links genomic data to the exact location of cells within tissues, combining high-resolution imaging with sequencing to map gene expression in context. This approach reveals how cells interact within their microenvironment, helping to understand tumor diversity, immune responses, and development. By preserving spatial information, researchers can identify unique biomarkers and cell communication patterns that bulk or single-cell methods miss.
Whole Transcriptome Sequencing (WTS)
RNA-Seq (RNA sequencing) is a high-throughput sequencing technique used to analyze the transcriptome, which is the complete set of RNA molecules transcribed in a cell. It allows for the comprehensive study of gene expression, alternative splicing, and other RNA-related phenomena.
Iso-Seq leverages PacBio long-read sequencing to sequence complete RNA transcripts without assembly. This provides unbiased, precise discovery of alternative splicing, novel isoforms, and fusion genes, offering a comprehensive view of isoform diversity for advanced research in cancer, neuroscience, and development.
LibSeq
Have libraries already prepared for various seqeuncing applications? Submit your pre-made libraries for rapid sequencing. We perform essential quality checks, process your samples, and deliver the results as raw data or with comprehensive bioinformatic analysis.

Isolation of DNA\RNA
We deliver high-throughput, automated nucleic acid extraction services, providing high-quality DNA and RNA from any sample type to accelerate your research.
Sanger Sequencing
Sanger Sequencing is a classical method of DNA sequencing developed by Frederick Sanger in 1977. It is also known as the chain-termination method and is widely used for accurate, small-scale DNA sequencing (e.g., verifying cloned DNA, detecting mutations, sequencing plasmids or PCR products).